
Log In
Log InSign up
Registration is for professionals that work with containment and Isolation Technologies and grants full access to our product data sheets and catalogues
Sign up

by Comecer Regenerative Medicine Solution Staff
The mass production of Advanced Therapy products continues to pose a challenge that has only been partially resolved, and which continues to evolve. Faced with the extreme variability inherent to the very concept of cell procedures, the operator’s task is to create the necessary conditions for stable growth in an effort to guarantee the procedure’s reproducibility and robustness. While a sufficient degree of success has been achieved in single procedures, the transition from an individual culture system to mass production is far from simple, given that the demand for increased volumes translates into the need for additional classified spaces, often impossible to create and in any case extremely costly. What’s more, a commercial approach brings to light the need for efficiency, which translates into an increasing focus on flexibility, modularity, and a reduction in costs. The proposed solution consists in an integrated modular system (isolator + incubator) for aseptic manipulation and cell culture expansion.
The system meets the strictest safety requirements; it is modular, expandable and with a view towards mass production, offers a viable solution to the cleanroom concept, significantly reducing production costs for Advanced Therapy products.
The theme of Regenerative Medicine, and therefore topics such as Tissue Engineering, Gene Therapy, Immunotherapy, Stem Cells and the like, are increasingly becoming the object of research, engineering and production, having surpassed the boundaries of those directly involved to become an issue of public interest, albeit with differing degrees of acceptance.
For those that work in the sector, all the above-listed topics can be summarised with the acronym ATMP (Advanced Therapies Medicinal Products) or rather medicinal products belonging to so-called “Advanced Therapies”, governed and subject to European legislation, specifically “Annex 1 and 2” of Good Manufacturing Practices (GMP). In fact, since 2007, ATMP have been compared to traditional pharmaceuticals (Regulation 1394/2007 EU) will all producers of sterile pharmaceuticals required to comply with said regulations, certifying the compliance of their products in order to obtain authorisation for their commercialisation within the European Union.
In short, an ATMP consists in a cell formulation, originating from the withdrawal of human tissue. It is likened to a pharmaceutical insofar as the final scope is its therapeutic administration to the patient. Cell formulations are classified as: autologous, in the case of formulations where the donor and recipient are the same person, heterologous in cases where the donor and recipient are different people. Legislation differentiates between an ATMP and a transplant, which assumes the withdrawal of an organ without any manipulation, and which is therefore regulated by ad hoc legislation. The road to the commercialisation of an ATMP is long and complex and involves a series of requisites that must be met by Advanced Therapy producers in order to obtain such authorisation. Many have worked and are working towards this goal, achieving gratifying results above all in the case of autologous treatments. However, the problems involved in the mass production of these medicines go well beyond their initial insertion into the market. In particular, the situation is radically different for small and large production scenarios, and while important results and successful, reproducible procedures have been developed for individual cases, the mass production of this medical technology continues to pose a number of difficulties.
As already highlighted in a previous article by the same authors (Sterility and Cross Contamination Challenges in Tissue Engineering and Regenerative Medicine), the production of ATMPs falls under the category of sterile pharmaceutical manufacturing and is subject to the same legislation. Regardless of the law, even non-experts are aware that any pharmaceutical of cellular origin, whose origins lie in a tissue biopsy and which is destined to be re-implanted in a patient, must be sterile. It is also clear that, given its “living” nature, an ATMP cannot be sterilised merely for the “production process” but rather must remain sterile for the entire duration of its manipulation, until its implantation in the patient. The issue is even more delicate for two reasons in particular: firstly, the peculiarity and uniqueness of the formulation (1 batch = 1 patient), and secondly the potential risk of the spread of contaminants in the culture environment, which by definition is a breeding ground for cell growth.
The objective of the researcher/lab technician is to develop procedures that can be reproduced in line with a predefined protocol, as accurately as possible. In manufacturing procedures based on a formula with established ingredients, reproducibility is easily achievable; however this is not the case for cell cultures. Although the protocol is the same, the unique characteristics of each individual mean that results may differ from expectations, even when “surrounding” conditions are identical. Hence the conclusion that in order for reproducibility to be maintained at the highest level (albeit not guaranteed), the culture conditions must be as constant as possible.
The robustness of a procedure implies its ability to resist against perturbations while maintaining its integrity. If the procedure is robust, its path will not easily be altered by any perturbations. Even if the process flow is altered, it will swiftly return to the predefined working conditions.
In the case of cell cultures, this implies the creation of stable “surrounding” conditions, the implementation of mechanisms that guarantee a rapid return to initial conditions in the case of perturbations, and where possible, prevention so as to minimise potential perturbations.
The obligatory question is: are national health systems able to cater to the demand for new therapies, and above all, can they afford it? This scenario clearly highlights the true challenge of ATMPs: the economic sustainability of managing and administering the therapy itself.
In this scenario, three different macro problems can be identified from both an operational and practical point of view:
In reality, there are three more issues that need to be addressed, but for the purposes of this analysis we will concentrate on those that appear to be particularly arduous problems to overcome. By analysing what we believe to be the current limitations, we will seek to understand whether there are any technological solutions, which when applied to the issues in question, may be able to simplify the road to the mass distribution of these therapies, encouraging their accessibility by a large number of patients, guaranteeing the quality of the process and reducing production costs.
The road to an ATMP pharmaceutical begins in a research laboratory, where the therapy is perfected. The laboratory is where initial hypotheses are made, the first tests are carried out, and often in vitro experiments and in vivo ones on test subjects. This is without a doubt a pioneering phase during which lab technicians normally work with a laminar flow hood, where the most critical manipulations in terms of sterility are carried out. Moreover, this phase involves the predominantly manual use of incubators, laboratory centrifuges, perishables, along with a large quantity of disinfectant sprays to decontaminate the working area and instruments.
Once a procedure is proven to be efficient in the pre-clinical phase, it must be prepared for the subsequent clinical study, typically with Phase I toxicity classification testing. To produce a series of “batches” (this notion is somewhat forced, given that these are autologous medicines, but it gives the right idea) production needs to be transferred to a sterile production workshop authorised by EMA (European Medicine Agency and AIFA – Agenzia Italiana del Farmaco – in Italy), or to a cleanroom or equivalent structure. It is in this structure that production will be authorised according to a specific procedure, subject to very strict monitoring rules and periodic validation.
In a sterile environment, biological samples are manipulated, expanded in special incubators, centrifuged, counted and prepared, normally for their re-infusion into the patient. This preparation is performed in accordance with the strictest sterility regulations, guaranteeing the quality of the product. These regulations, which are summarised in the attached table, are complied with in an environment organised as per Figure 1, which in fact represents a typical cleanroom.
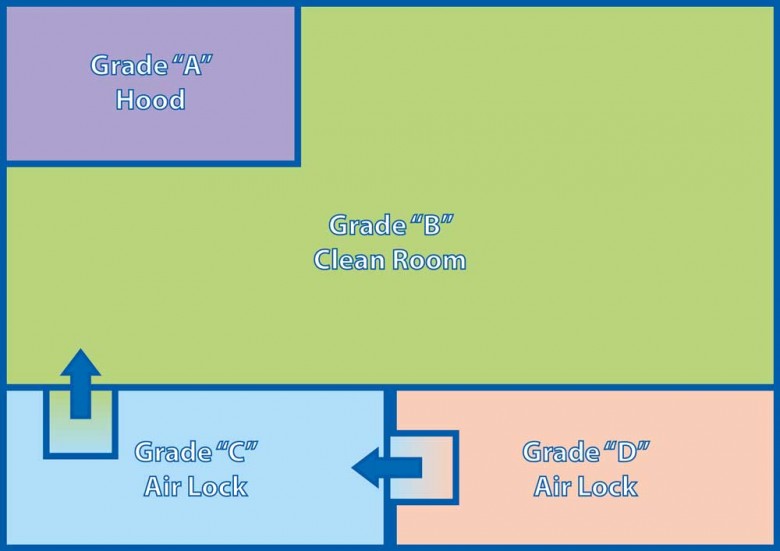
All instrumentation inside the cleanroom is held in grade “B” areas, while open-air manipulations occur under the laminar flow hood (represented by the grade “A” area in figure 1) where it is expected that the formulation will come into contact with the environment.
| Maximum permitted numbers of particles per m3 equal to or greater than the tabulated size | ||||
| At rest | In operation | |||
| Grade | 0.5 µm | 5 µm | 0.5 µm | 5 µm |
| A | 3 520 | 20 | 3 520 | 20 |
| B | 3 520 | 29 | 352 000 | 2 900 |
| C | 352 000 | 2 900 | 3 520 000 | 29 000 |
| D | 3 520 000 | 29 000 | Not defined | Not defined |
In its current state, the production of ATMPs in accordance with the proposed layout requires each cleanroom to be used for a specific product and dedicated exclusively to a specific application.
The incubator is the heart of the culture. As we know, the incubator is an environment where temperature and humidity are controlled and held constant; a mixture of air conditioned with CO2 is supplied in order to reproduce the physiological conditions for cell proliferation. During the period of incubation and depending on the application, the cultures preserved in the incubator are repeatedly accessed for observation, sampling, the addition of culture materials etc. Each time the culture needs to be accessed by operators, the latter access the grade “B” area in sterile clothing, open the incubator (and therefore it is opened in area “B”) and move the cultures underneath the laminar flow hoods in grade “A”, where they perform the necessary manipulations.
The possibility to regularly expand the production capacity of the laboratory is quite minimal insofar as the space classified as “grade B” by law is already entirely occupied. One possibility may be to add a few incubation stations, which in any case would lead to the saturation of the space in a very short time. Moreover, the additional equipment needs to be located in a classified area and making changes in such conditions is no easy task; it requires resources, time, and economic investments. A site’s production capacity is in fact normally expanded through the physical expansion of the surface area occupied by the classified areas.
At the end of a procedure, the preparation of a batch, a patient session, and even periodically, the incubator needs to be “re-cleaned”, as does the cleanroom, along with the decontamination of all surfaces, machines and materials used. The best incubators available in the market are equipped with a vapour sterilisation system, which while guaranteeing totally acceptable results in terms of quality, unfortunately mean that the incubator is inoperative for a number of hours (often more than 10 hours) while also impacting on the environmental conditions of the cleanroom (heating). The cleanroom is then cleaned in a sterile manner by dedicated workers in the same working conditions as for certified operators.
Isolation technology, in accordance with Annex 1 of the GMP, involves the creation of environments that are completely separated from the operator, inside which particle and microbiological cleanliness is maintained at grade “A”.
In accordance with the above-mentioned regulations, an isolator may be located in a grade “D” area, allowing operators to move in an area with decidedly lower limitations than grade “A” and “B” cleanroom classifications.
Some time ago COMECER developed an isolator specifically dedicated to cell culture activities (Fig. 2).

Fig. 2 Cell culture isolator
This isolator incorporates all the main devices (incubator, centrifuge, microscope, filling and refrigeration devices, and storage materials), allowing operators to perform operations normally carried out inside a cleanroom, entirely within the isolator without any contamination and contact with the outside environment, continuously maintaining grade “A” classification. Operator movements are minimal, while the cultures are exposed to grade “A” only and are not transported except for short distances within the isolator itself. Working times are therefore reduced while production efficiency increases. When necessary, incubators can be sterilised at the same time as the isolators in which they are incorporated, through a vapour-phase hydrogen peroxide (VPHP) sterilisation cycle, considered the Gold Standard of sterilisation in the biological sector.
These unique features of the isolator and the subsequent considerations provide a response to two of the major highlighted problems, specifically the efficiency and minimisation of movement, and the efficiency and affordability of the sterilisation process, however they do little in terms of the flexibility, modularity of installations and expandability of existing configurations.

The system consists in a series of modules, whereby each module represents a cell culture incubator, integrated into a Docking Station (Fig. 4) through which they share the utilities needed to operate (electrical power, humidified hot air, CO2, a decontamination system etc.).

Fig. 4 Flexycult Docking Station
The system is available in configurations with 3, 6, or 9 modules.
The incubators are sterilised through a vapour-phase hydrogen peroxide cycle before the start of the procedure, and are fed by an integrated HEPA 14 filtering system, which guarantees a constant grade “A” classification.
In order to manipulate the content, the incubator is then coupled to the isolator through a sterile safe RTP connection (Fig. 5). The incubators are therefore only opened inside an atmosphere with a guaranteed grade “A” classification.

Fig. 5 RTP connection
As the cell culture incubator respects the same conditions as the isolator, given that for all intents and purposes it communicates with the outside only in a grade “A” environment, it can be located in a grade “D” environment, in accordance with the GMP. This approach therefore leads to significant savings in terms of environment classifications. The procedure for connection-disconnection, use and return to position is described in Fig. 6.

Fig. 6 Sequence of use
At this point, having eliminated the limitation of needing to classify the environment in which the incubators are inserted, expansion no longer represents a major problem. To the contrary, where needed, a series of modules can be added in an area with a lower classification, with decidedly increased operating flexibility, optimising costs, logistics and personnel.
Fluidics management is centralised: the mixture of air, aqueous vapour and CO2 is prepared and pre-heated, then inserted into the incubator through a horizontal unidirectional flow
(Fig. 7) that guarantees the mixture’s consistent delivery and therefore its temperature, relative humidity and CO2 concentration. The presence of a horizontal unidirectional flow helps keep the environment clean (Fig. 8).

Fig 7 Homogenous air flow speed

Fig 8 Homogenous air flow direction
All transitions and the most important parameters are monitored and remotely controlled from a digital station (Fig. 9), which ensures maximum traceability of operations, contents and environmental conditions, guaranteeing supervision of the process and a prompt reaction in the event of a deviation in the procedure.
Fig 9 Monitoring and control interface
The following table summarises the key characteristics of the Flexycult™ product:
| Characteristic | Advantage | Benefit |
| Modularity | Progressive expandability | Improved capital and space management |
| Configuration flexibility | Improved space management | |
| VPHP sterilisation | Gold Standard sterilisation and automated cleaning procedure | Time saving and maximum efficiency |
| Humidified Air Delivery | No internal water tank | Improved cleaning, accuracy and protection against contamination |
| Centralisation of Fluids | Only 1 utilities connection for a certain number of incubators | Less wires, more linear control |
| Laminar flow and Grade A | Cleaner management. No operator-product contact. | Reduced risk of decontamination |
| Opening in isolator | Continuity of grade A | Reduced risk |
| Sealed environment | Possibility for location in grade D | Reduction of classified area |
| Wall heating | Fast incubation recovery time | Perfect temperature consistency and stable environmental conditions |
| Single-batch modules | No cross-contamination | Reduced risk |
| Digital Connection | Maximum monitoring level | Continuous control even by remote and prompt reaction |
Log in to your Comecer Account to download data sheets
Log In Lost PasswordNews and invitations to events and fairs directly in your mailbox
Subscribe